

Avoid this issues when starting a cooling tower
Conductivity controllers are critical devices in cooling tower systems, but they can develop issues when the tower has been dry for several months.
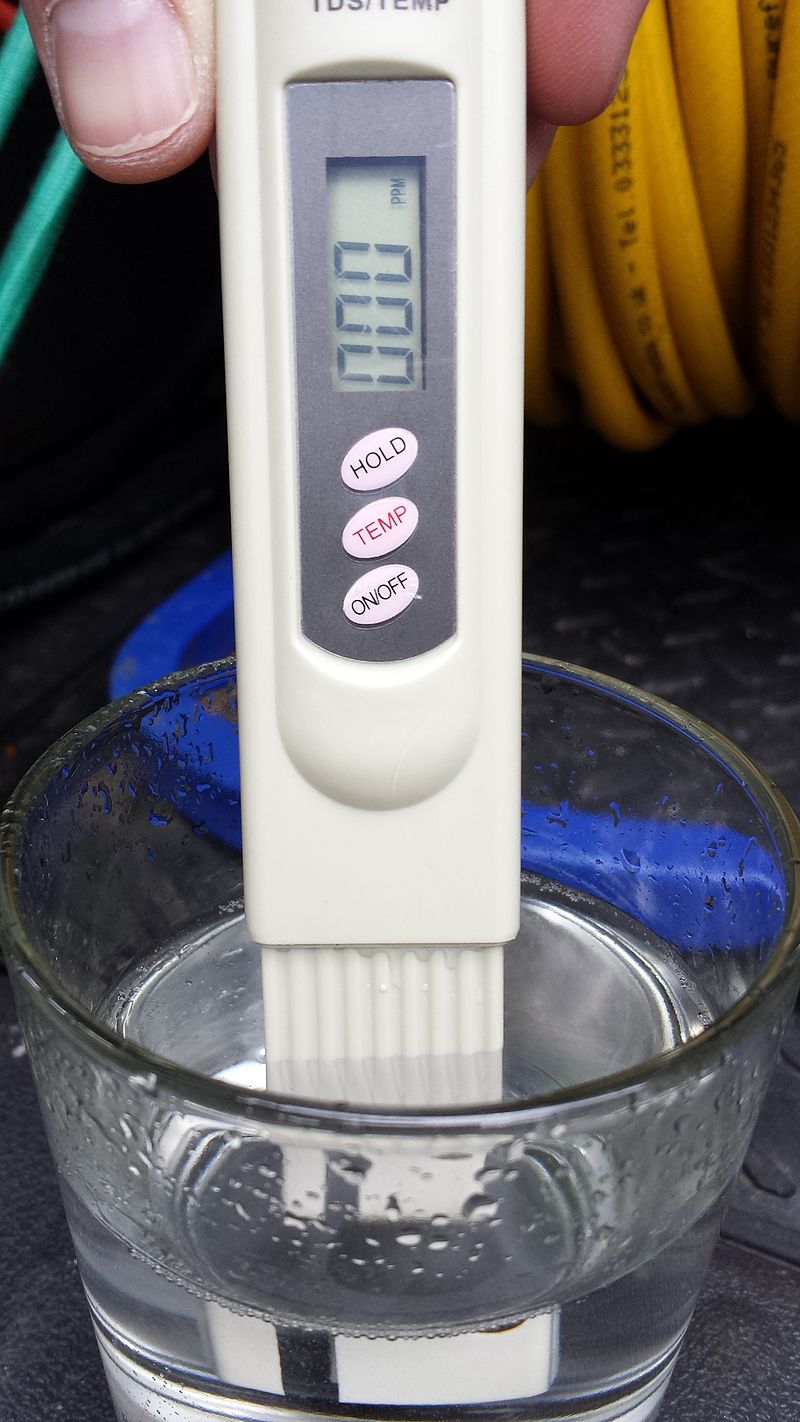
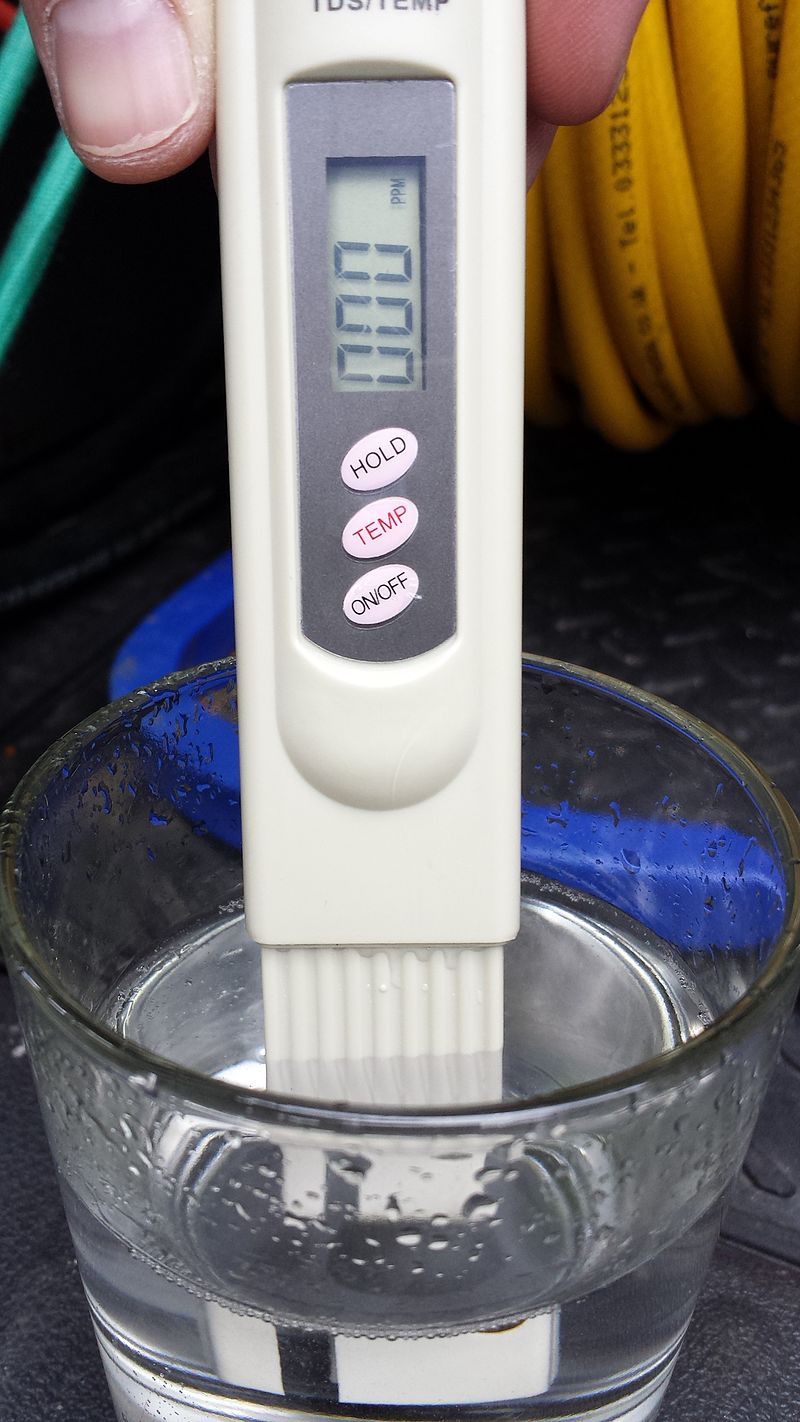
What is water TDS?
Total dissolved solids (TDS) is a measure of the dissolved combined content of all inorganic and organic substances present in a liquid in molecular, ionized, or micro-granular (colloidal sol) suspended form. TDS concentrations are often reported in parts per million (ppm). Water TDS concentrations can be determined using a digital meter.


Boiling point of water
Boiling occurs when the vapor pressure of a liquid equals the air pressure of the atmosphere above the liquid. For example, at sea level, water boils at 212°F (100°C). As elevation increases, the amount of atmosphere above the liquid decreases, so the boiling temperature of the liquid decreases. In general, the lower the atmospheric pressure, the lower the boiling temperature of any liquid.




Sulfites for Oxygen Control
The sulfite/oxygen reaction is known to be inhibited by some alcohols, phenols, amines, and thiosulfate. Other contaminants or organic treatment chemical such as corrosion inhibitors, scale inhibitors, and biocides may also slow down reaction time. A slow reaction can present a problem at early phases in a system and require the use of catalysts or feeding techniques that provide maximum time for the reaction to occur. The reaction rate for sulfite appears to be the fastest of all of the scavengers, followed by erythorbic acid and DEHA. Slower rates, in general, have been reported for hydroquinone, carbohydrazide, and hydrazine.










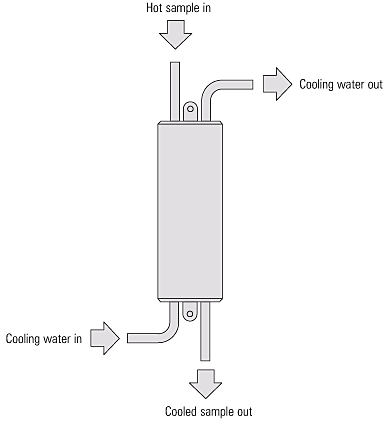

Fundamentos de la Medicion de la Conductividad de Calderas
Mientras una caldera genera vapor, impurezas que están en el agua de alimentación de la caldera y, que no son arrastradas por el vapor generado, se concentrarán en el agua de la caldera. Dentro de la caldera el calor genera burbujas de vapor dentro del agua. Estas burbujas flotan y se rompen al llegar a la superficie d...


